What is Yeast Genetics?
The advantage of using Saccharomyces cerevisiae in Yeast Genetics
Why is Saccharomyces cerevisiae such a good model organism?
Saccharomyces cerevisiae has been the model organism for most of the molecular genetics research in the past two decades for a number of reasons (1):
- cerevisiae can stably exist in both haploid and diploid states. This allows scientists to easily isolate recessive mutants and map out genes that are responsible for a particular phenotype.
- Foreign DNA in the form of plasmids or linear nucleotides can easily be transformed in cerevisiae. This has allowed scientists to conduct genetic complementation analysis and other engineering techniques (See point 3).
- cerevisiae has a strong homologous recombination pathway and a high rate of gene conversion. Combined with the development of DNA transformation techniques, scientists can directly integrate exogenous DNA sequences into desired chromosomal loci, creating mutant strains with different markers.
- There are hundreds of genetic markers available in cerevisiae (See below), which allow scientists to easily generate stable mutant strains (Point 3) or trace multiple markers simultaneously.
- Since the genome sequence of cerevisiae is known, the science community has generated many genome-wide library collections of strains and plasmids. These collections are invaluable tools for scientists to perform genome-wide, large-scale genetics and genomics screens.
Genetic Markers for Saccharomyces cerevisiae
The nutritional requirements for yeast growth have been clearly defined over the years, and the pathways for synthesizing these requirements have been discovered (2). If a biochemical step in one of these pathways is disrupted, yeast will no longer survive unless the missing nutrient is supplied in the growth media.
A wild-type yeast cell that has the ability to synthesize its own nutritional requirement is called a prototroph. Its mutant counterpart that loses the ability to synthesize an essential nutrient due to a DNA mutation somewhere in the biosynthetic pathway is called an auxotroph. An auxotrophic marker is then defined as a wild-type allele of a gene that encodes a key enzyme for the production of an essential monomer used in biosynthesis,
As a result, scientists can track hundreds of auxotrophic markers by simply changing the composition of the growth media. This characteristic has made yeast the most powerful genetic tool of the 20th century! Some examples of the commonly used auxotrophic markers in S. cerevisiae are URA3, LYS2, LEU2, TRI1, HIS3, MET15 and ADE2 (1). All these marker genes encode essential enzymes for de novo nucleic acid and amino acid synthesis. Two immediate applications for yeast auxotrophic markers are the stable maintenance of expression vectors and the introduction of knockout mutations.
Genetic Analyses in Saccharomyces cerevisiae
In general, scientists use three genetic techniques to isolate and characterize unknown mutations in S. cerevisiae. These techniques are molecular cloning, genetic complementation, and tetrad dissection (1,2).
In molecular cloning, a library of single-copy vectors containing inserts of random genomic fragments is transformed into unknown mutant strains (Figure 1). The transformants are examined for the non-mutant (Yfg+) trait. Once the transformant with the desired phenotypes is identified, the vector is isolated. The identity of the random sequence on the vector can be further narrowed down by restriction digestion or identified by sequencing. Molecular cloning is particularly useful if there are no available mutant strains that can be crossed to.
Genetic complementation can be used to reveal the identity of an unknown mutation if this mutant exhibits the same phenotype as other mutant strains with a known identity (Figure 2). Specifically, an unknown mutation in a haploid strain is crossed with a wild-type haploid strain of opposite mating type, as well as a set of haploid strains that exhibit the same phenotype. The diploid crosses are isolated and the unknown mutant phenotype is scored. The mutation is recessive if the heterozygous control diploids do not exhibit a mutant phenotype. The diploid cross that exhibits the same mutant phenotype would indicate that the unknown mutation is the same as the known mutant that was initially crossed.
Tetrad dissection can be used to identify the genetic linkage of a mutation and to determine if the unknown mutation is an alternation at a single genetic locus (Figure 3). In tetrad analysis, the haploid unknown mutant is crossed with a wild-type haploid strain or a strain with known genetic markers. The diploid cross is isolated and induced for sporulation. The resulting four spores from the same meiotic event are separated and scored for the desired phenotype. A 2:2 segregation pattern would indicate that the phenotype is the result of a single gene. Analyzing segregation patterns could reveal genetic linkages to other markers or centromere.
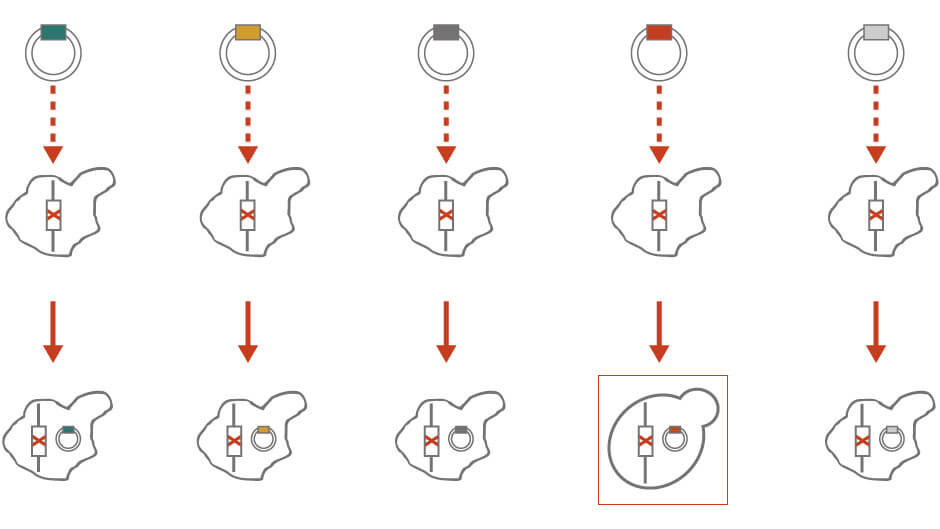
Figure 1: Molecular cloning for genetic analysis. See text for details.
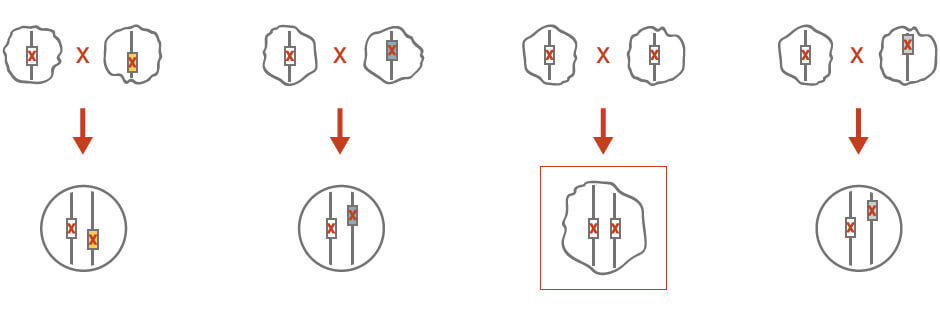
Figure 2: Gene complementation for genetic analysis. See text for details.
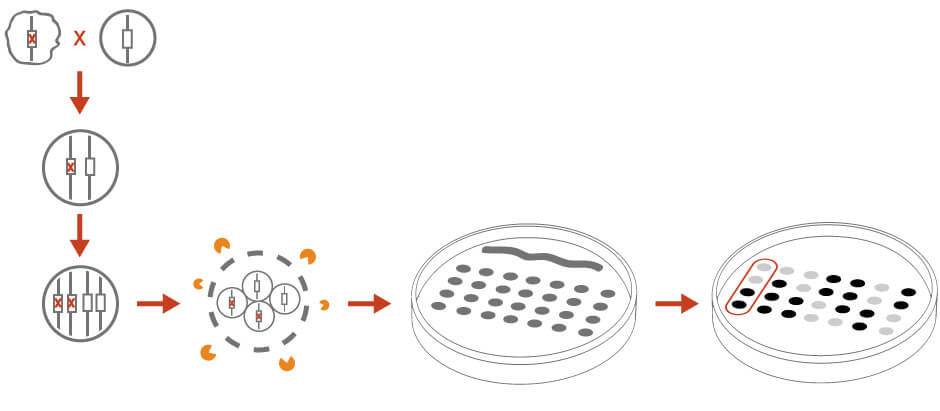
Figure 3: Tetrad analysis for genetic analysis. See text for details.
Want to understand how geneticists map genes in yeast?
Our next article reveals the power of tetrad dissection.