Next generation tools for high throughput genome editing in yeast
Donor-templated CRISPR gene editing
Deciphering natural variation in yeast
Kevin Roy is a research scientist at Stanford Genome Technology Center, whose research is fascinating to us here at Singer Instruments. With deep-rooted expertise in yeast genetics and molecular biology, Kevin’s work is a vibrant mix of cutting-edge high-throughput sequencing and sophisticated data analysis. His passion lies in exploring the intricate interactions between genetic variation and the environment, unraveling how this complex interplay shapes cellular phenotypes.
In this article, we’re showcasing MAGESTIC, a powerful tool Kevin and his team developed to decipher the significance of natural variation in yeast. Singer Instruments’ PIXL and ROTOR+ systems have been instrumental in this work, enabling precise colony picking, efficient library generation, and high-throughput phenotypic screening.
By integrating these automation tools into his workflow, Kevin has been able to scale his screening capacity from daily runs of six or seven hours to fully-walkaway, overnight picking bonanzas arraying up to 15,000 colonies at time.

The science of complex traits
Variation is the essence of biology: it fuels adaptation and evolution. For complex traits involving many genes, it can be challenging to distinguish the contribution of all the small genomic differences (e.g. Single Nucleotide Polymorphisms, SNPs) between two individuals when those individuals have so many differences.
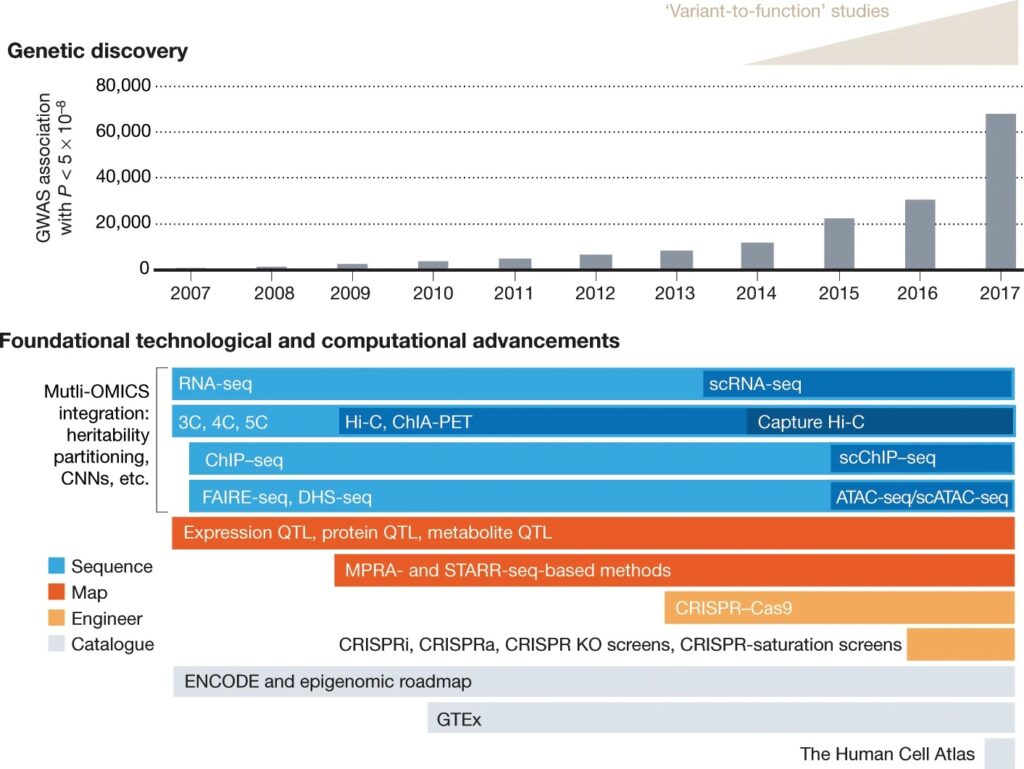
While the list of GWAS associated loci continues to grow (Fig. 1), each one can harbour dozens to thousands of SNPs and it’s a challenge to identify which are truly causal. If it were possible to make precise changes to a genome and then observe how these changes affect the organism, it would revolutionise the way these genetic variants can be characterised. And this is exactly what Kevin and his team have been working on.
CRISPR + Homology-Directed Repair — It’s precise but not efficient
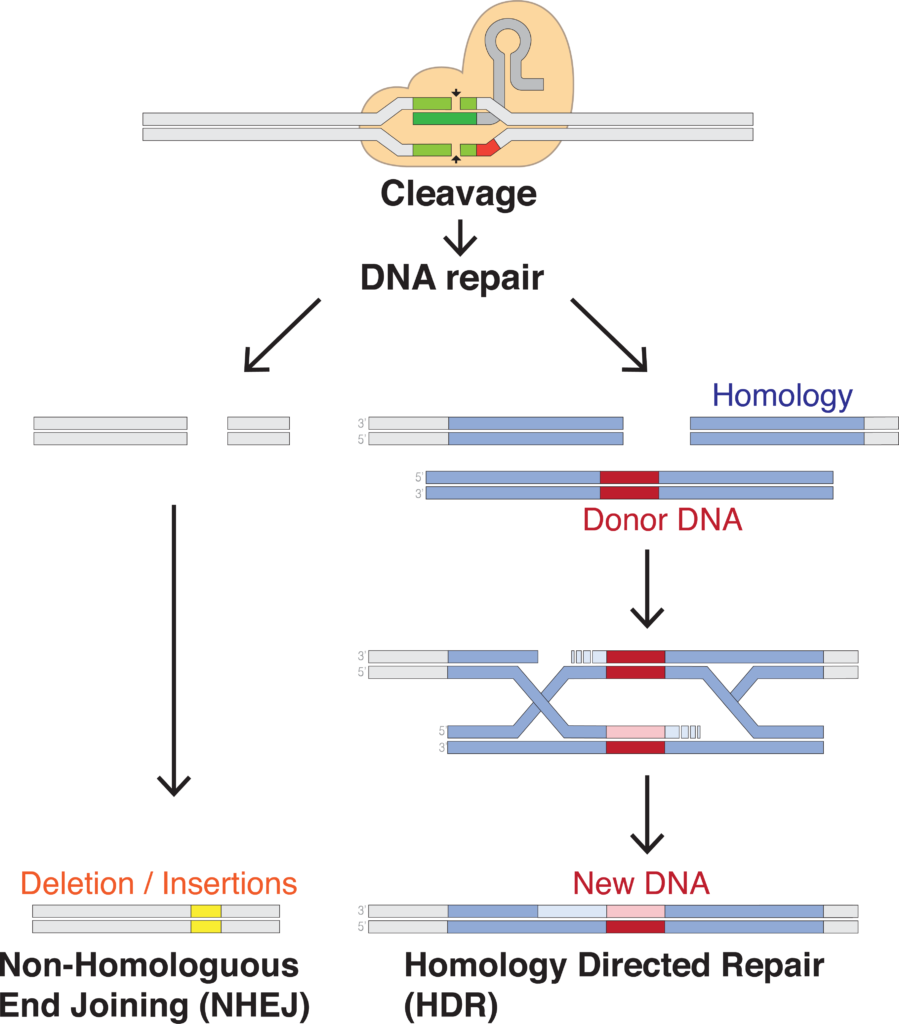
CRISPR is a revolutionary tool that leverages a cell’s machinery to make specific edits nearly anywhere in the genome. These breaks are fatal to cells if left unchecked, meaning the cellular machinery scrambles to fix them in one of two ways (Fig. 2).
In the DNA repair mechanism Non-Homologous End Joining (NHEJ), the cell repairs the break by sticking the ends back together — not always perfectly — which introduces random variation, often insertions and deletions of nucleotides (indels). This randomness can be useful in some applications, but when you need to understand the impact of specific genetic changes, you need something more precise.
That’s why the DNA repair mechanism Homology-Directed Repair (HDR) is much more useful for dissecting natural variation. Unlike NHEJ, HDR uses a matching chromosome to fix the break accurately. This mechanism can be hijacked by providing a template with the desired mutations (the so-called donor DNA) which will then be faithfully integrated into the cut site.
While this sounds fantastic, applying HDR on a large scale to simulate natural variation is challenging. It’s a complex and inefficient process, especially when there are thousands of genetic variants to decipher. The bottlenecks are just too substantial to make this approach viable.
{kind=link}
A MAGESTIC solution
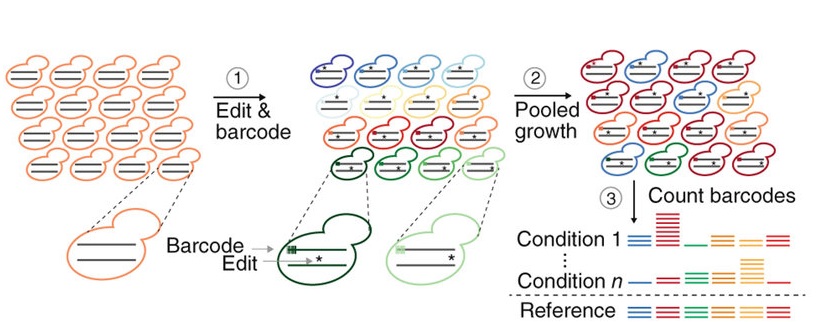
In 2018, Kevin and his team introduced MAGESTIC —Multiplexed Accurate Genome Editing with Short, Trackable, Integrated Cellular barcodes (fig. 3). The technique uses high throughput technology to make precise edits in the genome, using CRISPR + HDR to reflect natural variation.
A short genetic barcode is also integrated into the genome to make it easier to identify whether the edits are successful. But, that’s not all. Kevin’s team further developed an ingenious method to increase the efficiency of HDR by more than five-fold by improving donor DNA recruitment to the site of a double-strand break.
The team has since published a preprint paper to announce MAGESTIC 3.0. This version of MAGESTIC has new features to increase efficiency further still and reduce the frequency of pesky random mutations from being injected into the cut sites via NHEJ.
In short, MAGESTIC is a game-changer in systematic genome editing, making it possible to explore the vast landscape of genetic variation in yeast with unprecedented precision and efficiency.
Majestic robots: PIXL and ROTOR+
To leverage the full power of MAGESTIC, many tens of thousands of mutants need to be generated to build a comprehensive library representing natural variation. With a library this large, employing automation is the only effective way to screen for interesting phenotypes with enough precision and throughput.
Pooling together the genomes of 16 diverse yeast strains originated from the 1002 yeast genomes project, Kevin and his team are now using PIXL to create complex genetically-barcoded libraries that aim to catalogue the vast genetic diversity found in nature. Thanks to MAGESTIC, these have higher efficiency, fidelity, and superior variant representation than pre-existing donor-guided approaches.
Prior to implementing fully automated colony picking, Kevin’s team relied on a technician to create 384 arrays at a rate of around 5,000 colonies a week. Now with PIXL Momentum continuously picking by day or night and at 1536 densities, they are able to plate an average of 100,000 colonies a week, and have freed up their technician for other activities. In total, the team has successfully picked more than 1 million colonies with next to zero errors, a move that Kevin says has has enabled the team to “address previously unapproachable questions.”

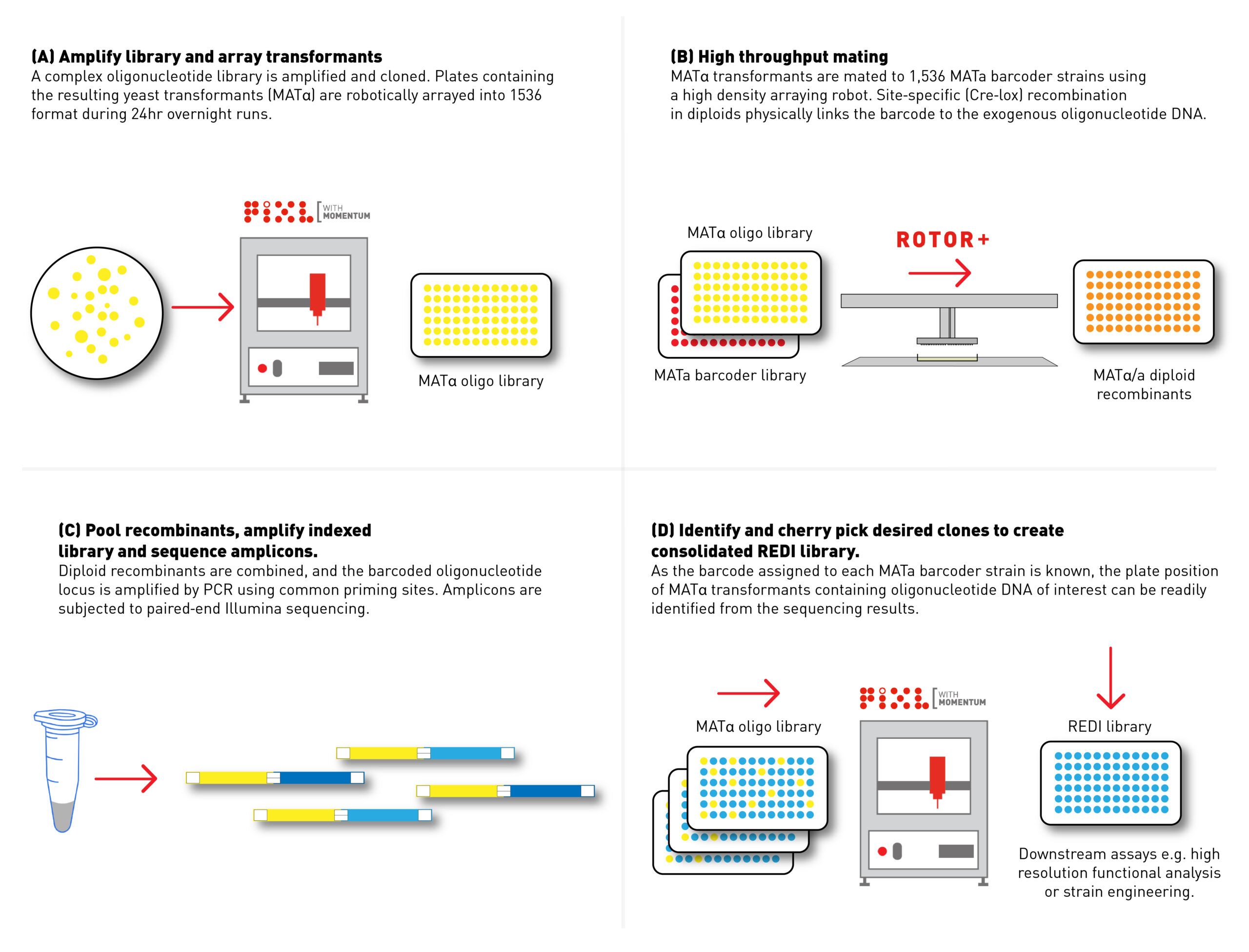
Fully automated colony picking. Are you REDI?
Using another innovative technique worked on by Kevin as part of an earlier project, called Recombinase Directed Indexing (REDI), his team are leveraging ROTOR+ and PIXL to create sublibraries that group together genes of interest. By first consolidating libraries, REDI helps minimize false positives—a common limitation in standard pooled methods caused by competition for phenotyping capacity. And since REDI is also amenable to automation, the method enables the rapid and cost-effective production of high-quality DNA libraries.
The combined might of MAGESTIC and REDI unlocks new possibilities for high-resolution functional analysis at throughputs previously thought unattainable. Kevin’s team is now applying this cutting-edge approach across various synthetic genomic applications, including identifying natural variants that drive phenotypic responses to heavy metals.
We at Singer Instruments are of course delighted to support this development and excited for the findings to be made available soon!
Impress your Supervisors with more details
Register for our On-Demand webinar with Dr Kevin Roy on high-throughput genome editing with MAGESTIC 3.0.

Phil Kirk PhD | Senior Scientist
Phil heads up our Research team, combining his biological knowledge, programming experience and engineering skills to push our robots to their limits and leading experimental work to back up their use in a variety of applications and settings.
He has a background in plant science and biotechnology, and close to a decade of laboratory experience. Hence, he’s as happy wielding a micropipette as a screwdriver, loves cracking complex scientific problems, and is an absolute whizz at designing an R-script!
By ensuring our innovations do what they are designed to do, Phil’s work contributes to more reliable robots, meaning more time our customers can spend performing the work that can’t be automated: creating, interpreting, and enjoying science!